
The human transcriptome across tissues and individuals
During my postdoc at the CRG, I participated in the Genotype-Tissue Expression project (GTEx). This project was aimed at finding genetic variants that affected gene expression (eQTLs). Within the project, 1641 samples from 43 tissues coming from 175 individuals were analyzed using RNA-seq. This dataset represented a unique opportunity to study how gene expression varied across tissues and individuals at an unprecedented scale and so we did! Below I summarize some of the main findings of the study:
We were interested in studying in depth how expression and splicing varied across tissues, and also we wanted to identify genes that were highly variable across groups such as males and females, different ethnicities and age that could explain differences in disease susceptibility.
We first looked at the proportion of the variance in gene expression that could be explained by tissue and by individual. We found that a significant proportion of the variation in gene expression could be explained by variation across tissues rather individuals and that this observation was true both for lncRNAs and for protein coding genes.
We then looked for genes that varied between males and females, between European Americans and African Americans and across different ages. We saw that genes that varied with ethnicity were enriched in lncRNAs, whereas genes that varied with age were mostly protein coding. In addition, by merging information on GWAS we are able to identify specific candidate genes that could explain differences in susceptibility to cardiovascular diseases between males and females and different ethnic groups. Finally, we found that genes that decrease with age were involved in neurodegenerative diseases such as Parkinson and Alzheimer and identified novel candidates that could be involved in these diseases.
In contrast to gene expression, we found that splicing varied similarly among tissues and individuals, and exhibited a larger proportion of residual unexplained variance. This may reflect that stochastic, non-functional fluctuations of the relative abundances of splice isoforms may be more common than random fluctuations of gene expression. However, comparatively, the contribution of individual variation is larger for splicing (10%) than for expression (4%). This re-inforced the idea that whereas gene expression is a more important determinant of the cellular phenotype than splicing, splicing, may play an important role in modulating phenotypic differences between individuals.
Non-coding RNA evolution
Evolutionary history of human populations
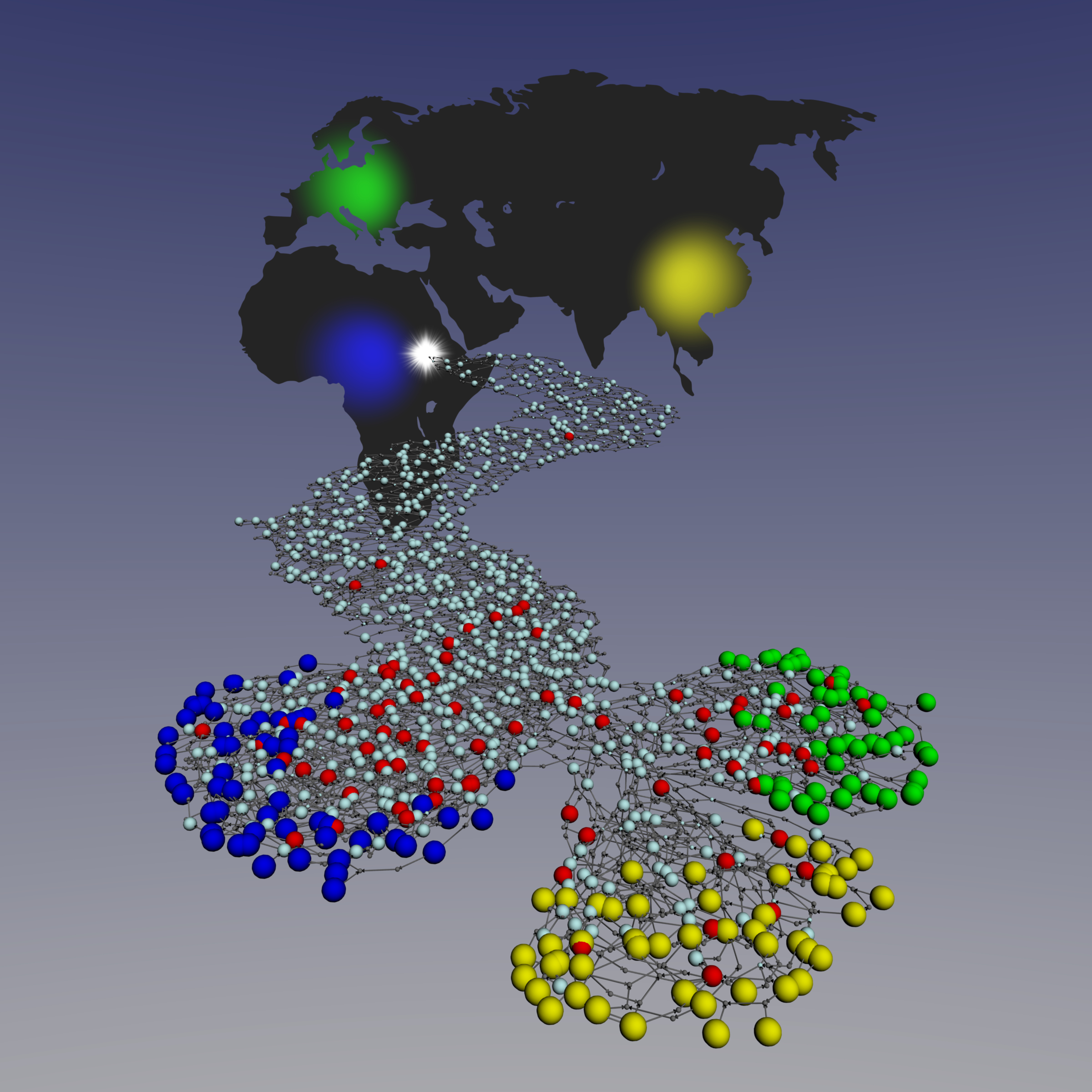
Simulated coalescent network representing the Out of Africa event and the emergence of three human groups: Africans (blue), Europeans (green) and Asians (yellow). Light blue nodes represent past recombination events and small gray nodes represent coalescent events. Red nodes are past recombination events recovered by IRiS. These coalescent networks were used to fine-tune and validate IRiS. Coalescent network were generated with cosi (Shaffner et al. 2005) and displayed using Pajek software . Figure by Marc Pybus.
The aim my PhD was to use the information left by recombination in our genomes to make inferences on the recent evolutionary history of human populations. For that, we developed a novel method called IRiS that allowed detecting specific past recombination events in a set of extant human sequences. IRiS was extensively validated and studied in whole set of different scenarios in order to assess its performance. Once recombination events are detected, they can be used as genetic markers to study the recombinational diversity patterns of human populations.
We applied this innovative approach to a set of human sequences from several populations of the Old World that were specifically genotyped for this end. We found evidence that anatomically modern humans most likely left Africa through the Bab-el-Mandeb strait rather than through present Egypt. In addition, we estimated the effective population size of all these populations and found that sub-Saharan African populations had four times larger effective size than non-African populations and that South Asian populations had the largest effective population of the out-of-Africa populations.